
近日,药学院徐云根/朱启华/邹毅团队在药物化学顶尖期刊Journal of Medicinal Chemistry发表最新研究论文:Rational Design of PARP1/c-Met Dual Inhibitors for Overcoming PARP1 Inhibitor Resistance Induced by c Met Overexpression。药学院博士研究生孙泽人、李兰杰和硕士研究生翟冰新为本论文的共同第一作者,邹毅副研究员、徐云根教授和朱启华教授为本文的共同通讯作者,中国药科大学为本文的唯一通讯单位。
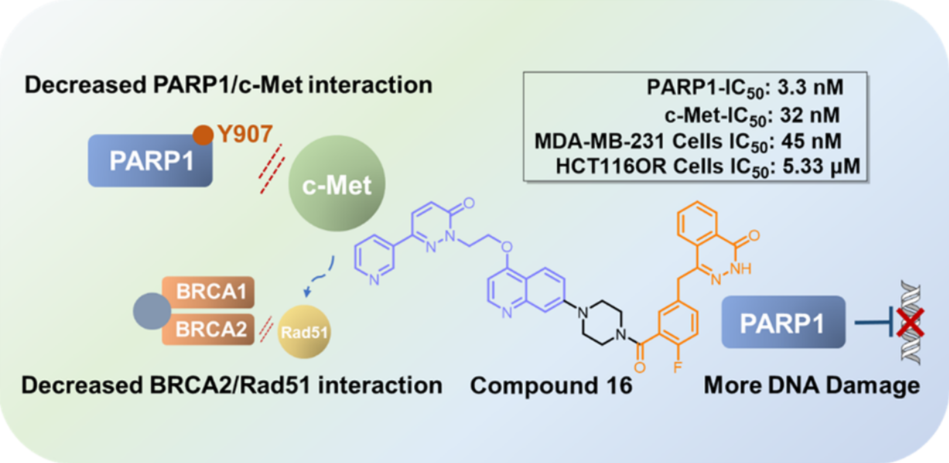
PARP1抑制剂是肿瘤精准治疗的代表性药物,目前已有6款PARP1抑制剂被批准上市用于携带BRCA1/2基因缺失或突变的乳腺癌、卵巢癌等肿瘤的治疗。虽然PARP1抑制剂在临床抗肿瘤治疗中取得了巨大的成功,然而大部分患者在长期使用后仍不可避免地产生耐药性(获得性耐药);此外,仍有40%携带BRCA1/2基因缺失或突变的肿瘤患者对PARP1抑制剂不敏感(原发性耐药性)。研究发现,c-Met扩增的肿瘤细胞可分别通过磷酸化PARP1和Rad51,导致肿瘤细胞对PARP1抑制剂产生原发性耐药和获得性耐药。说明c-Met是PARP1抑制剂应答的关键调节因子,PARP抑制剂和c-Met抑制剂同时使用,可以改善c-Met扩增的肿瘤患者对PARP1抑制剂的耐药性。基于这些发现,该团队以PARP1抑制剂Olaparib和c-Met抑制剂化合物1为先导化合物,通过合理药物设计,设计合成了一系列喹啉类衍生物。体内外活性筛选发现了优势化合物16,该化合物对两个靶点均具有较好的抑制活性(PARP1 IC50: 3.3 nM;c-Met IC50: 33.2 nM),且在BRCA突变型与野生型肿瘤细胞中均表现出优于Olaparib的抗增殖效果。值得注意的是,化合物16对c-Met扩增的BRCA野生型肿瘤细胞MDA-MB-231增殖的抑制活性(IC50 = 45 nM)是Olaparib(IC50 = 30.7 μM)的680倍,其对Olaparib诱导的获得性耐药细胞HTC116OR增殖的抑制活性(IC50 = 5.33 μM)是Olaparib(IC50 = 50.8 μM)的10倍。进一步体内药效评估表明,不管肿瘤细胞是否发生BRCA1/2突变,化合物16均能够有效抑制肿瘤的生长,并且显著优于Olaparib。总之,该团队开发出了首款PARP1/c-Met双靶点抑制剂,为解决由于c-Met扩增导致的PARP1抑制剂耐药性提供了新的方案。
此外,该团队分别于2020和2021年在Journal of Medicinal Chemistry发表了关于PARP/PI3K双靶点抑制剂(J. Med. Chem. 2020, 63, 122-139.)和PARP/BRD4双靶点抑制剂(J. Med. Chem. 2021, 64, 17413-17435.)的研究成果,旨在通过不同靶点与PARP1的协同机制增强PARP1抑制剂的抗肿瘤作用、拓宽PARP1抑制剂的适应症。
以上工作获得江苏省自然科学基金(No. BK20211220)和国家大学生创新创业训练计划(No. 202310316009Z)的资助。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00077
示意图
(供稿单位:药学院,撰写人:刘华)